List of publications:
Google Scholar ID: 6CT8AGIAAAAJ
Web of Science ResearcherID: ABA-3331-2021
MTMT ID: 10032702
Our programs are regularly released in the many-purpose MRCC quantum chemistry program package open-access for academic use.
Development of highly-accurate and affordable quantum chemistry methods:
We are facing ever more complicated chemical questions both from the perspective of interpretation and design of new experiments, as well as of their understanding and modeling at the atomic scale. Our team focuses on the latter by developing predictive quantum chemical methods and their more and more affordable implementations.
We are at the forefront of the development and application of quantum chemistry's gold standard [so called coupled cluster, CCSD(T)] method, that has been repeatedly found to provide chemical accuracy compared to experiments, that is sufficient accuracy for chemical reactions and molecular interactions.
Our recent, efficiently paralellized[1], reduced-cost[2], and especially the reduced-scaling[3,4] and local natural orbital (LNO) based CCSD(T) codes[5,6,7] extended the reach of chemically accurate modeling up to record-sized molecules (of 100s or even a 1000 atoms).[7,8] This efficiency is achieved via systematically controllable physics- and numerical maths-based approximations combined with efficient algorithms designed for modern high-performance computing.
Our latest LNO-CCSD(T) implementation has a number of particularly practical features enabling uniquely accurate and efficient computations for large molecular systems with routinely accessible hardware.[7,8] Namely, both the closed and open-shell[4] implementations are highly optimized for low operation count and memory requirement, with negligible disk and network use. To control the precision, we suggested a systematically convergent LNO approximation hierarchy suitable for extrapolation and introduced conservative LNO error estimates.

Further unique features of LNO-CCSD(T) include, frequent checkpointing, and treatments of point group symmetry and near-linear dependent AO basis sets. The LNO approach is also available for general order CC and second-order excited state methods:

We also extensively benchmarked the precision of the LNO approximations on reactions and interactions of realistic molecules[6,7,19]. Moreover, all independent comparisons so far showed that the LNO-CCSD(T) method provides highly competitive accuracy and efficiency for reaction and interaction energies, e.g., for
- organic thermochemistry,[9,10]
- non-covalent interactions,[11,12]
- as well as for organometallic-[13,14] and extended π-systems [9,15] exhibiting even moderate strong correlation.
Accurate modeling of molecular interactions and reactions:
Exploiting the systematic convergence of he LNO and AO basis set hierarchies, it is relatively affordable to approach the basis set limit of CCSD(T) for molecules of 100s of atoms. For example, we investigated
- orgnacatalytic reaction mechanisms [16],
- the stability of phosphinyl radicals[17], as well as
- various ion-ligand[18], organic dimer[19], and protein-ligand[7] interactions.
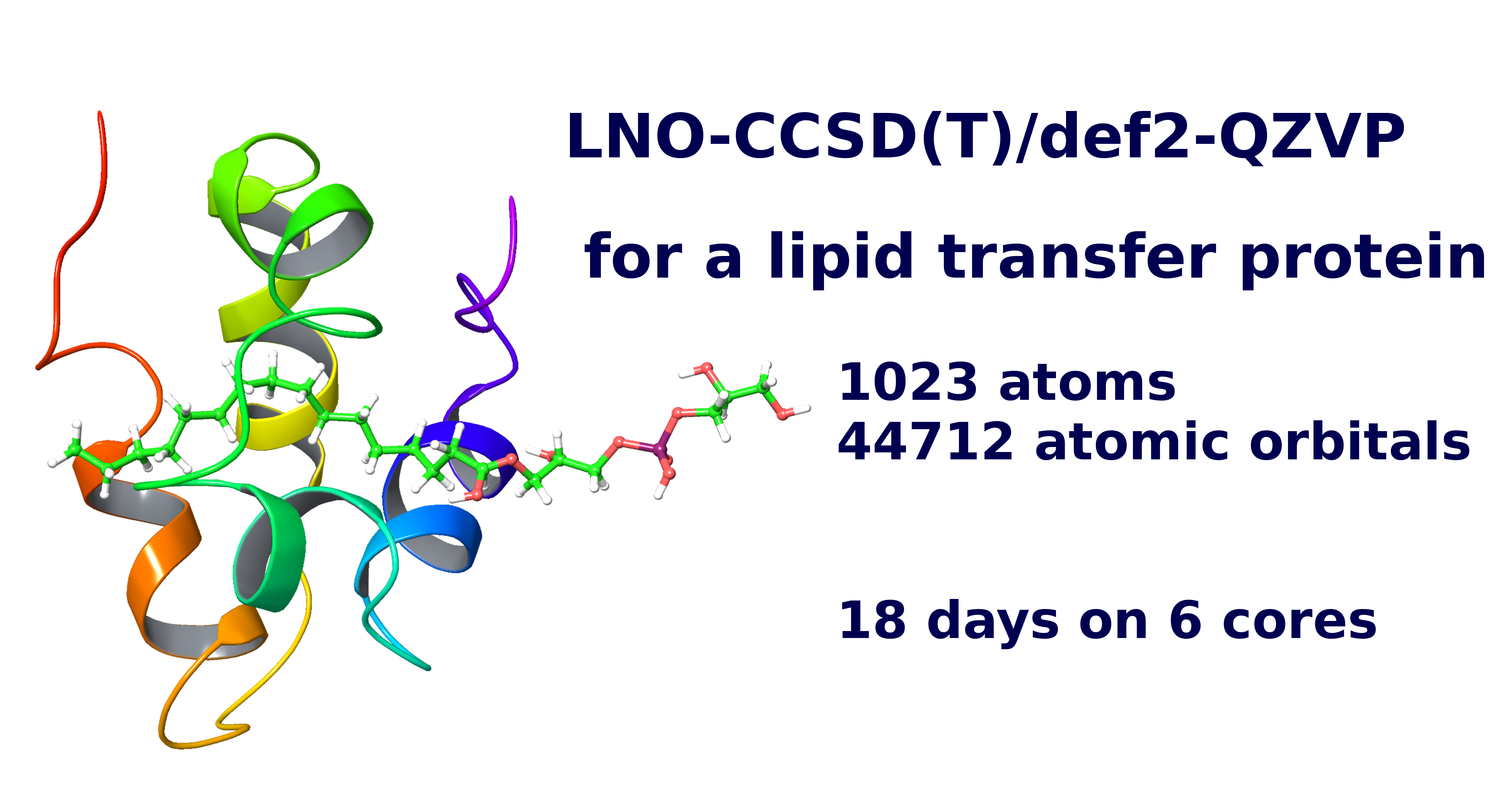
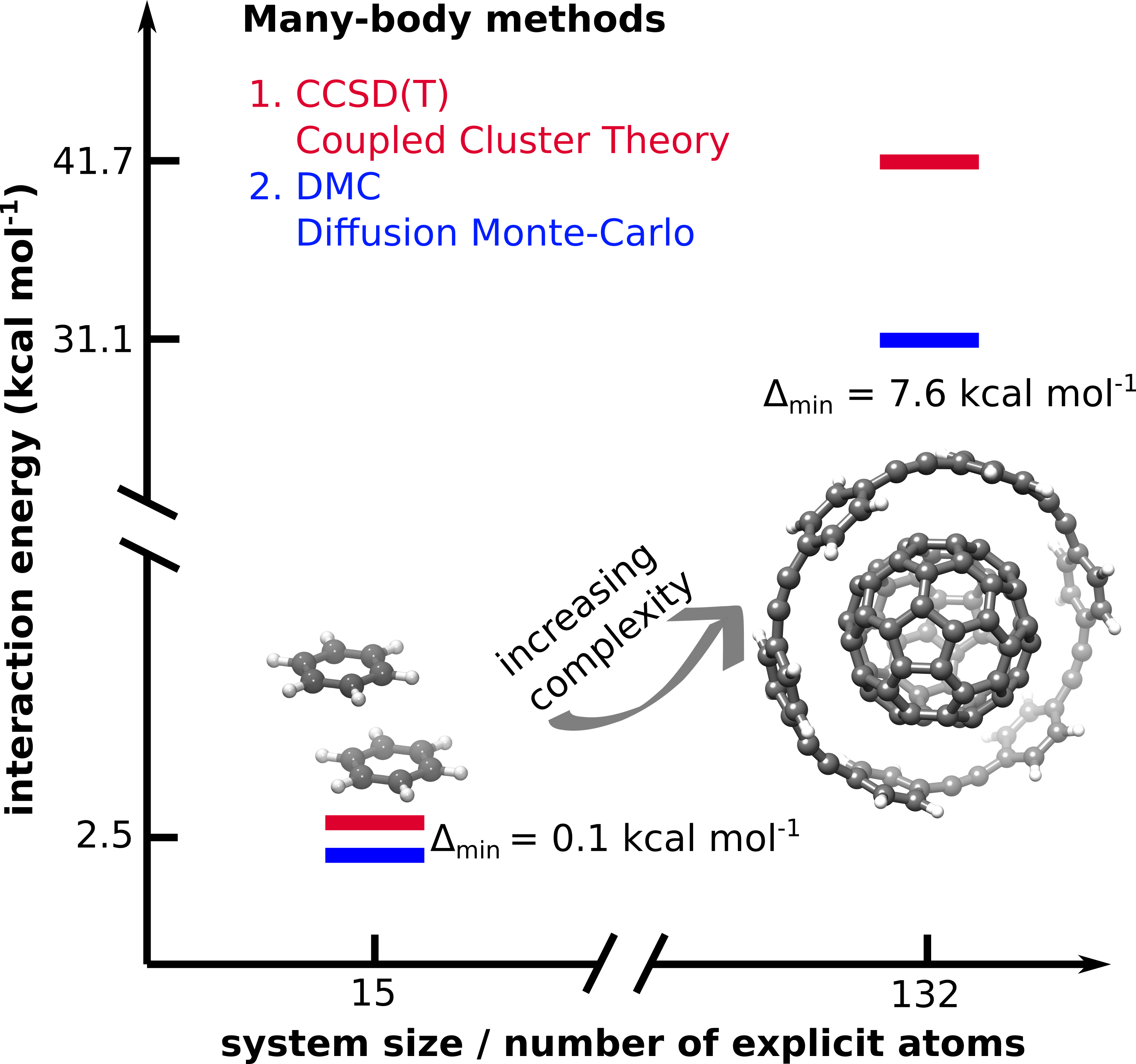
Our well-converged CCSD(T)-level supramolecular interaction energies generally also agree excellently with similarly highly-regarded fixed-node diffusion Monte Carlo (FN-DMC) results.[8] However, in an exciting collaboration with the group of Alexandre Tkatchenko, Yasmine Al-Hamdani, Gerit Brandenburg, and Andrea Zen, we found that the two reference methods deviate well outside their estimated error bars for a few polarizable molecular complexes, which puzzling result is still under investigation...