Applications from motivated and qualified colleagues in undergraduate, Ph.D. or postdoc stages are welcome at any time.
Additionally, we fully encourage and support applications and proposal writing for individual or joint funding via Marie Skłodowska-Curie Actions, Erasmus, Fulbright, Stipendium Hungaricum, or other mobility fellowships, European Research Council grants, Hungarian Research Fund, Hungarian Academy of Sciences, ÚNKP, etc. grants and fellowships.
We are also eligible for resources allocated to EU widening countries, e.g., the European Research Area (ERA) fellowship increases the chances of winning MSCA postdoc fellowships with us by ca. 40%!
Participation in consortial projects are also welcomed.
We also participate in international short/mid-term mobility and visiting scholarships. You can visit and collaborate with us upon contacting us for invitation or via scholarships offered in multiple counties both in and out of Europe (e.g., Argentina, Australia, Brazil, Canada, China, India, Korea, Mexico, Singapore, South Africa, Thailand, United States, Romania, Croatia, Cyprus, Estonia, Italy, Republic of Serbia, Slovak Republic, Slovenia), SIRE program in India, Swiss Postdoc.Mobility.
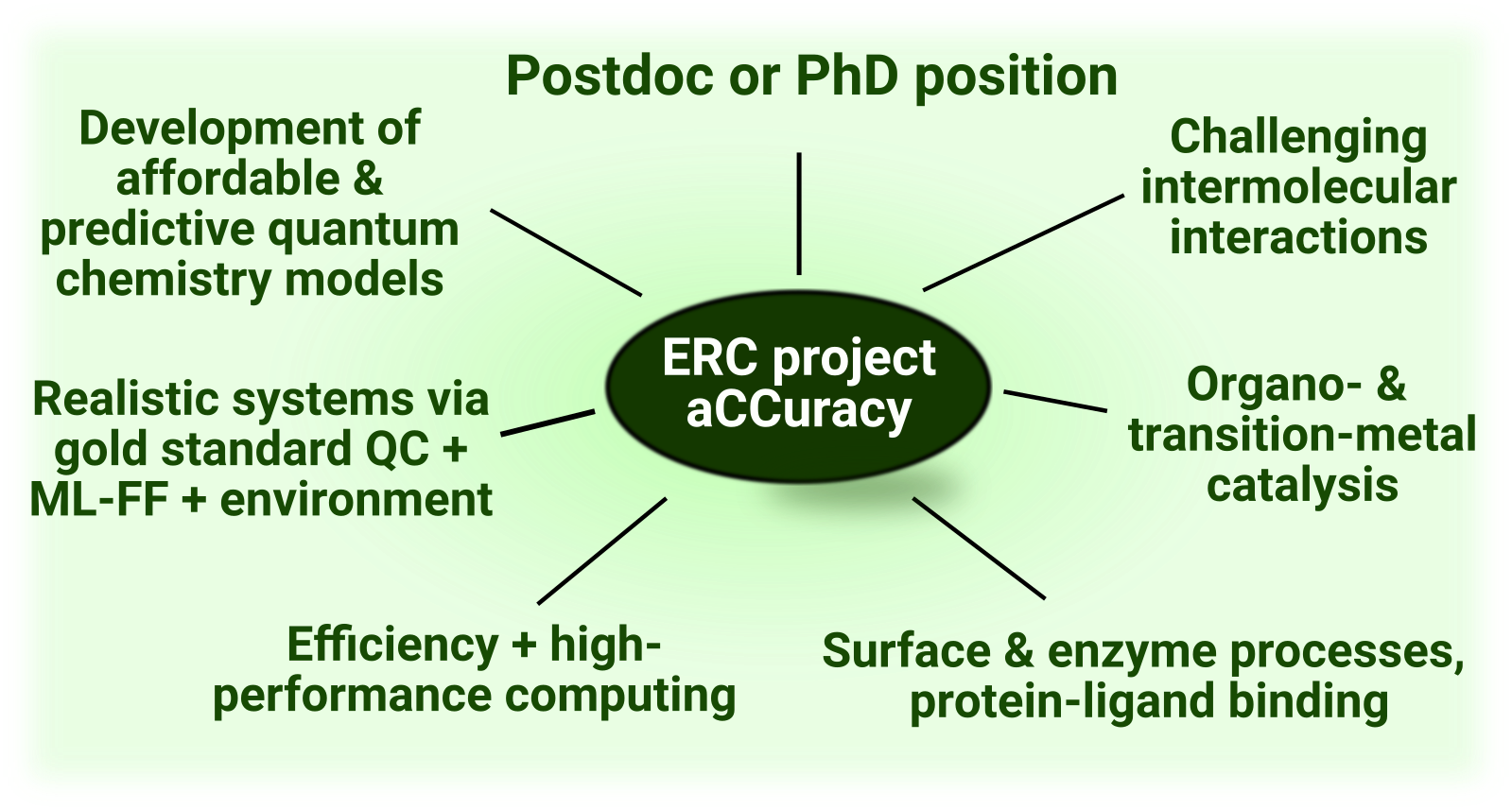
Currently, Ph.D. and postdoc positions are available funded via our ERC Starting grant project.
Ph.D. and undergraduate students
- with Chemistry, Physics, Material Science, Computer Science or related background and
- with experience and extensive motivation in theoretical/computational modeling of reactions and molecular interactions and/or
development/programming of quantum chemistry models
are encouraged to look at the project descriptions below and contact Péter to discuss potential thesis projects. Projects are available in the Oláh György PhD School and application for international students are also encouraged, e.g., via Stipendium Hungaricum or other fellowships.
Postdoc positions
Project description:
We aim at further improving the accuracy, efficiency, and functionality of our cutting-edge conventional and local electron correlation based electronic structure models, especially at the many-body perturbation (PT) theory and coupled cluster (CC) level, as well as at the intersection of DFT and wave function based approaches. This is achieved via concerted high-performance software design, as well as theoretical and algorithmic developments in our local natural orbital (LNO) family of methods.
For example, while the accuracy of the gold standard CCSD(T) model has been repeatedly corroborated against experiments, our accelerated CCSD(T) approaches became one of the most efficient variants, extending the reach of chemically accurate modeling up to record-sized molecules (of 100s or even a 1000 atoms) [1,2,3]. Our open-access programs [4,5] are already used in many research groups worldwide and were repeatedly found to be among the most efficient and accurate by all independent comparisons. The developments are part of the MRCC program package with 1000+ registered users.
Successful candidates will contribute to some of the development (D) projects:
D1) The accuracy and speed of our methods will be substantially increased by implementing our new ideas for better approximations (via, e.g., higher-order perturbative estimates, explicit electron correlation, improved long-range interactions, etc.) and a massively parallel code suitable for use in the largest supercomputers.
D2) Development and practical implementation of similarly efficient DFT and local CCSD(T) level observables, such as thermodynamic, structural, spectroscopic, and dynamic molecular properties.
D3) Further development and application of our multilevel or embedding methods using gold standard accuracy for the chemically active region combined with cost-efficient models (MP2, DFT, ML, MM) to take into account biochemical, crystal, and solvent environment effects.
D4) Blending wavefunction and DFT methods to extend and improve our double-hybrid and wavefunction-in-DFT embedding approaches.
D5) Extensions of these efficient and accurate DFT, PT, and CC methods to open-shell and multireference systems.
These ongoing developments enable the uniquely reliable simulation of intricate chemical processes of practical importance, which are both complicated to study experimentally and not accessible with chemical accuracy via any other lower-cost model. In particular, our aim is the predictive modeling and atomistic understanding of challenging covalent- and non-covalent interactions in large molecules, where modern workhorse computational methods (such as DFT) have well-known difficulties:
1) complicated long-range aromatic, ionic, and hydrogen/halogen-bond interactions between large molecules governing, e.g., supramolecular and catalyst-substrate interactions. E.g., protein-drug interactions.
2) the mechanism of environment-friendly and selective organo-, and transition-metal catalytic reactions involving both closed- and open-shell species. E.g., CO2 and N2 reduction with earth-abundant d-block metal catalysis.
3) surface and enzyme catalysis, as well as protein-drug interactions via multi-level embedding models to take into account solvent, ionic crystal, and protein environment effects. E.g., computational mechanistic study of phosphate reactions relevant to cancer formation or human DNA and viral RNA polymerization and related drug desin.
[1] Chemical Science 15, 14556 (2024)
[2] Journal of Chemical Theory and Computation 15, 5275 (2019) & 17, 860 (2021)
[3] Nature Communications 12, 3927 (2021), J. Am. Chem. Soc. 139, 17052 (2017)
[4] J. Chem. Phys. 152, 074107 (2020)
[5] J. Phys. Chem. A 129, 2086 (2025)
Your qualifications:
- PhD in Chemistry or Physics or Material Science or Computer Science or Data Science or related fields with solid research experience in ab initio molecular/materials modeling (electronic structure methods) OR numerical simulations OR machine learning OR scientific programming in related fields
- ideally strong method development/programming experience with wave function or DFT based models related to our research goals OR extensive experience with machine learning force fields and dynamics simulations
- ideally strong scientific programming skills (in FORTRAN/C or related and in a scripting language) and high-performance computing experience
- strong publication record compared to your career stage and background
- motivation and experience to conduct independent research. The PI and the MRCC developer team provides guidance, support in frequent consulations
- ability to contribute to the activities and duties of our research team: minimal amount of teaching in the host institution (1-2 hours/week/person) and potentially (co-)supervision of (under)graduate students as well as participation in collaborations and application projects
- ideally experience with or potential/plan to apply for research grants/scholarships appropriate to your career stage
- good written and oral communication skills in English, ideally proven ability to draft quality research papers/proposals. The working language is English (knowledge of Hungarian language is not necessary).
We offer:
- research ideas and projects at the forefront of predictive atomistic simulations, working with our group, collaborators, and the MRCC developer team
- exposure to physicists, chemists, programmers, and engineering mindsets
- frequent consultations, mentoring, and support proportional to your invested effort
- distribution of developed code to increasing MRCC user base and proper code co-authorship credit
- encouragement and support to develop your own research ideas, apply for scholarships/fellowships, and work toward your scientific goals
- outstanding salary corresponding to your carrier stage, especially considering the standard of academic positions and the cost of living in the host country (health and pension benefits are also included)
- full time, one year research position with potential extensions up to five years in case of mutual interest (ideally at least 2-3 year postdoc projects are preferred tackling a larger goal)
- flexible working hours and partial home office is a possibility
- BME is a leading university in the country, located on a beautiful World Heritage site in one of the most affordable capitals of the European Union
Application instructions:
Please, send to Péter via e-mail in a single pdf/doc file with subject line “ERC job application FAMILY NAME” the following:
- short cover letter describing your research experience, strengths/weaknesses in the above qualifications, preferred start date interval (available immediately or later upon mutual agreement) and research topic(s) of your interest
- current CV, publication list, scientific indicators (preferably including a link to Google Scholar/ ResearcherID/etc. publication profile)
- contact detail of and your relation described to at least two researchers in the field who can be asked for a letter of recommendation in the case your application is shortlisted
Feel free to contact Péter via e-mail for more details and questions before application.